Properties: Density of States (DOSS)¶
The CryDossCalculation can be used to run the properties
executable for DOSS calculations, from an existing fort.9.
See also
Properties Workflow to run multiple properties calculations (and optional initial SCF).
!verdi plugin list aiida.calculations crystal17.doss
Inputs
code: required Code The Code to use for this job.
parameters: required Dict the input parameters to create the properties input file.
wf_folder: required FolderData, RemoteData, SinglefileData the folder containing the wavefunction fort.9 file
metadata: optional
Outputs
remote_folder: required RemoteData Input files necessary to run the process will be stored in this folder node ...
results: required Dict Summary Data extracted from the output file(s)
retrieved: required FolderData Files that are retrieved by the daemon will be stored in this node. By defa ...
arrays: optional ArrayData energies and DoS arrays
Exit codes
1: The process has failed with an unspecified error.
2: The process failed with legacy failure mode.
10: The process returned an invalid output.
11: The process did not register a required output.
200: The retrieved folder data node could not be accessed.
210: The main (stdout) output file was not found
211: The temporary retrieved folder was not found
300: An error was flagged trying to parse the crystal exec stdout file
350: The input file could not be read by crystal
351: Crystal could not find the required wavefunction file
352: Parser could not find the output isovalue (fort.25) file
353: Error parsing output isovalue (fort.25) file
400: The calculation stopped prematurely because it ran out of walltime.
401: The calculation stopped prematurely because it ran out of memory.
402: The calculation stopped prematurely because it ran out of virtual memory.
413: An error encountered usually during geometry optimisation
414: An error was encountered during an scf computation
415: An unknown error was encountered, causing the mpi to abort
499: The main crystal output file flagged an unhandled error
from aiida import engine, load_profile, orm, plugins
from aiida.tools.visualization import Graph
from aiida_crystal17.common import recursive_round
from aiida_crystal17.tests.utils import (
get_or_create_local_computer, get_or_create_code,
get_default_metadata)
from aiida_crystal17.tests import open_resource_binary
import matplotlib.pyplot as plt
%matplotlib inline
profile = load_profile()
profile.name
'test_crystal17'
Running a calculation¶
The DOSS Input Schema gives the allowed format of the input dictionary, for example:
computer = get_or_create_local_computer('work_directory', 'localhost')
code = get_or_create_code('crystal17.doss', computer, 'mock_properties17')
builder = code.get_builder()
builder.metadata = get_default_metadata()
builder.parameters = orm.Dict(dict={
"k_points": [18, 36],
"npoints": 100,
"band_minimum": -10,
"band_maximum": 10,
"band_units": "eV"
})
with open_resource_binary('doss', 'mgo_sto3g_scf', 'fort.9') as handle:
builder.wf_folder = orm.SinglefileData(handle)
result, calcnode = engine.run_get_node(builder)
!verdi process show {calcnode.pk}
Property Value
------------- ------------------------------------
type CalcJobNode
pk 884
uuid ff55e9d9-9a03-4375-b5d9-2353794f1c92
label
description
ctime 2019-09-24 14:04:37.274585+00:00
mtime 2019-09-24 14:04:51.167679+00:00
process state Finished
exit status 0
computer [1] localhost
Inputs PK Type
---------- ---- --------------
code 881 Code
parameters 882 Dict
wf_folder 883 SinglefileData
Outputs PK Type
------------- ---- ----------
arrays 888 ArrayData
remote_folder 885 RemoteData
results 887 Dict
retrieved 886 FolderData
graph = Graph(graph_attr={'size': "6,8!", "rankdir": "LR"})
graph.add_node(calcnode)
graph.add_incoming(calcnode, annotate_links="both")
graph.add_outgoing(calcnode, annotate_links="both")
graph.graphviz

Analysing the outputs¶
The outputs are:
results a dict of computation input and output parameters, parsed from the stdout file.
arrays a set of energy and DoS arrays for each point
recursive_round(calcnode.outputs.results.get_dict(), 1)
{'newk': {'k_points': [18, 18, 18],
'gilat_net': 36,
'n_kpoints_ibz': 195,
'n_kpoints_gilat': 1240},
'npts': 102,
'spin': False,
'units': {'energy': 'eV', 'conversion': 'CODATA2014'},
'errors': [],
'header': {'crystal_version': 17, 'crystal_subversion': '1.0.1'},
'warnings': [],
'wf_input': {'n_ao': 14,
'n_atoms': 2,
'k_points': [8, 8, 8],
'n_shells': 5,
'n_symops': 48,
'gilat_net': 8,
'n_core_el': 12,
'n_electrons': 20,
'energy_fermi': -4.0,
'energy_total': -7380.2,
'n_kpoints_ibz': 29,
'energy_kinetic': 7269.0},
'energy_max': 10.4,
'energy_min': -10.2,
'system_type': 'closed shell, insulating system',
'fermi_energy': -4.0,
'parser_class': 'CryDossParser',
'parser_errors': [],
'parser_version': '0.11.0',
'norbitals_total': 14,
'parser_exceptions': [],
'execution_time_seconds': 0}
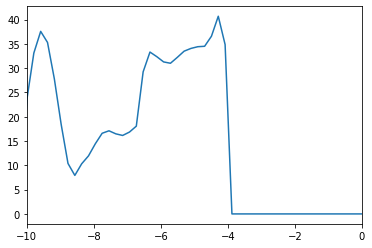
plt.plot(
calcnode.outputs.arrays.get_array("energies"),
calcnode.outputs.arrays.get_array("total"))
plt.gca().set_xbound(-10, 0)
Computing Projections¶
Projections can be added per atom or per orbital set.
Note
A maximum of 15 projections are allowed per calculation.
Note
orm.Dict(dict={
"shrink_is": 18,
"shrink_isp": 36,
"npoints": 100,
"band_minimum": -10,
"band_maximum": 10,
"band_units": "eV",
"atomic_projections": [0, 1],
"orbital_projections": [[1, 2, 3]]
})
<Dict: uuid: c2ec5f24-bd4b-46fe-ba21-23b121ad3f6d (unstored)>
In order to create orbital sets,
it is possible to compute the nature of each orbital,
using the atomic structure and basis sets used to create the fort.9:
from aiida_crystal17.tests import get_test_structure_and_symm
from aiida_crystal17.symmetry import print_structure
structure, _ = get_test_structure_and_symm('NiO_afm')
print_structure(structure)
StructureData Summary
Lattice
abc : 2.944 2.944 4.164
angles : 90.0 90.0 90.0
volume : 36.1
pbc : True True True
A : 2.944 0.0 0.0
B : 0.0 2.944 0.0
C : 0.0 0.0 4.164
Kind Symbols Position
---- ------- --------
Ni1 Ni 0.0 0.0 0.0
Ni2 Ni 1.472 1.472 2.082
O O 0.0 0.0 2.082
O O 1.472 1.472 0.0
basis_cls = plugins.DataFactory('crystal17.basisset')
basis_sets = basis_cls.get_basissets_from_structure(structure, 'sto3g')
basis_data = {k: v.get_data() for k, v in basis_sets.items()}
basis_data
{'Ni': {'type': 'all-electron',
'bs': [{'type': 'S', 'functions': ['STO-nG(nd) type 3-21G core shell']},
{'type': 'SP', 'functions': ['STO-nG(nd) type 3-21G core shell']},
{'type': 'SP', 'functions': ['STO-nG(nd) type 3-21G core shell']},
{'type': 'SP', 'functions': ['STO-nG(nd) type 3-21G core shell']},
{'type': 'D', 'functions': ['STO-nG(nd) type 3-21G core shell']}]},
'O': {'type': 'all-electron',
'bs': [{'type': 'S', 'functions': ['STO-nG(nd) type 3-21G core shell']},
{'type': 'SP', 'functions': ['STO-nG(nd) type 3-21G core shell']}]}}
from aiida_crystal17.parsers.raw.parse_bases import compute_orbitals
cresult = compute_orbitals(structure.get_ase().numbers, basis_data)
print("number of electrons: ", cresult.electrons)
print("number of core electrons: ", cresult.core_electrons)
cresult.ao_indices
number of electrons: 72
number of core electrons: 40
{1: {'atom': 0, 'element': 'Ni', 'type': 'S', 'index': 1},
2: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 1},
3: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 1},
4: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 1},
5: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 1},
6: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 2},
7: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 2},
8: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 2},
9: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 2},
10: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 3},
11: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 3},
12: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 3},
13: {'atom': 0, 'element': 'Ni', 'type': 'SP', 'index': 3},
14: {'atom': 0, 'element': 'Ni', 'type': 'D', 'index': 1},
15: {'atom': 0, 'element': 'Ni', 'type': 'D', 'index': 1},
16: {'atom': 0, 'element': 'Ni', 'type': 'D', 'index': 1},
17: {'atom': 0, 'element': 'Ni', 'type': 'D', 'index': 1},
18: {'atom': 0, 'element': 'Ni', 'type': 'D', 'index': 1},
19: {'atom': 1, 'element': 'Ni', 'type': 'S', 'index': 1},
20: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 1},
21: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 1},
22: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 1},
23: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 1},
24: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 2},
25: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 2},
26: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 2},
27: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 2},
28: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 3},
29: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 3},
30: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 3},
31: {'atom': 1, 'element': 'Ni', 'type': 'SP', 'index': 3},
32: {'atom': 1, 'element': 'Ni', 'type': 'D', 'index': 1},
33: {'atom': 1, 'element': 'Ni', 'type': 'D', 'index': 1},
34: {'atom': 1, 'element': 'Ni', 'type': 'D', 'index': 1},
35: {'atom': 1, 'element': 'Ni', 'type': 'D', 'index': 1},
36: {'atom': 1, 'element': 'Ni', 'type': 'D', 'index': 1},
37: {'atom': 2, 'element': 'O', 'type': 'S', 'index': 1},
38: {'atom': 2, 'element': 'O', 'type': 'SP', 'index': 1},
39: {'atom': 2, 'element': 'O', 'type': 'SP', 'index': 1},
40: {'atom': 2, 'element': 'O', 'type': 'SP', 'index': 1},
41: {'atom': 2, 'element': 'O', 'type': 'SP', 'index': 1},
42: {'atom': 3, 'element': 'O', 'type': 'S', 'index': 1},
43: {'atom': 3, 'element': 'O', 'type': 'SP', 'index': 1},
44: {'atom': 3, 'element': 'O', 'type': 'SP', 'index': 1},
45: {'atom': 3, 'element': 'O', 'type': 'SP', 'index': 1},
46: {'atom': 3, 'element': 'O', 'type': 'SP', 'index': 1}}
To observe DoS at the fermi level, these results can also be used to choose a sensible range of bands:
filled_bands = int(cresult.electrons / 2)
first_band = int(cresult.core_electrons / 2) + 1
last_band = min([first_band + 2 * (filled_bands - first_band), cresult.number_ao])
first_band, last_band
(21, 46)
O_sp_orbitals = [k for k, v in cresult.ao_indices.items()
if v["element"] == "O" and v["type"] == "SP"]
Ni_d_orbitals = [k for k, v in cresult.ao_indices.items()
if v["element"] == "Ni" and v["type"] == "D"]
computer = get_or_create_local_computer('work_directory', 'localhost')
code = get_or_create_code('crystal17.doss', computer, 'mock_properties17')
builder = code.get_builder()
builder.metadata = get_default_metadata()
builder.parameters = orm.Dict(dict={
"k_points": [18, 36],
"npoints": 1000,
"band_minimum": first_band,
"band_maximum": last_band,
"band_units": "bands",
"orbital_projections": [O_sp_orbitals, Ni_d_orbitals]
})
with open_resource_binary('doss', 'nio_sto3g_afm', 'fort.9') as handle:
builder.wf_folder = orm.SinglefileData(handle)
result2, calcnode2 = engine.run_get_node(builder)
calcnode2.outputs.arrays.attributes
{'array|energies': [1002],
'array|total_beta': [1002],
'array|total_alpha': [1002],
'array|projections_beta': [2, 1002],
'array|projections_alpha': [2, 1002]}
round(calcnode2.res.fermi_energy, 2)
6.89
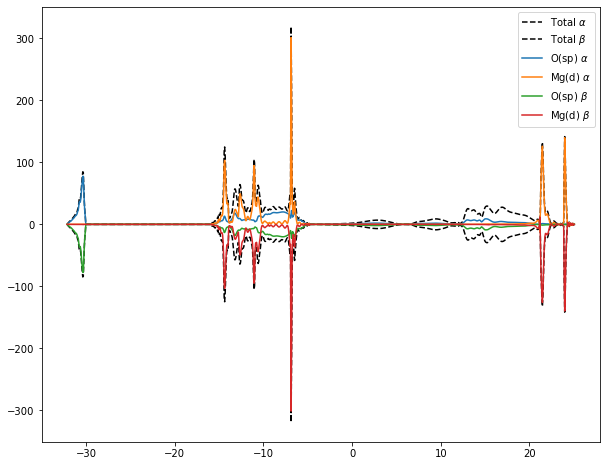
plt.plot(
calcnode2.outputs.arrays.get_array("energies"),
calcnode2.outputs.arrays.get_array("total_alpha"),
label="Total $\\alpha$",
color="black", linestyle="dashed"
)
plt.plot(
calcnode2.outputs.arrays.get_array("energies"),
calcnode2.outputs.arrays.get_array("total_beta"),
label="Total $\\beta$",
color="black", linestyle="dashed"
)
plt.plot(
calcnode2.outputs.arrays.get_array("energies"),
calcnode2.outputs.arrays.get_array("projections_alpha")[0],
label="O(sp) $\\alpha$"
)
plt.plot(
calcnode2.outputs.arrays.get_array("energies"),
calcnode2.outputs.arrays.get_array("projections_alpha")[1],
label="Mg(d) $\\alpha$"
)
plt.plot(
calcnode2.outputs.arrays.get_array("energies"),
calcnode2.outputs.arrays.get_array("projections_beta")[0],
label="O(sp) $\\beta$"
)
plt.plot(
calcnode2.outputs.arrays.get_array("energies"),
calcnode2.outputs.arrays.get_array("projections_beta")[1],
label="Mg(d) $\\beta$"
)
plt.legend();
plt.gcf().set_size_inches(10, 8)