This page was generated from docs/source/user_guide/calc_main.ipynb,
with configuration: sphinx_ipypublish_all.ext.noexec
Main Calculation Plugin¶
The crystal17.main plugin is the core calculation plugin. It is
designed with a more programmatic input interface , to create the input
.d12 and .gui files, from a set of AiiDA
Data nodes.
See also
See Main Calculation Immigration for a method
to immigrate
existing output/input files as a
crystal17.main calculation.
Initial Setup¶
To run a computation, first ensure AiiDA is running:
In:
!verdi status
✓ profile: On profile test_crystal17
✓ repository: /Users/cjs14/GitHub/aiida-cjs-working/databases/aiida/.aiida/repository/test_crystal17
✓ postgres: Connected as cjs14@localhost:5432
✓ rabbitmq: Connected to amqp://127.0.0.1?heartbeat=600
✓ daemon: Daemon is running as PID 35477 since 2019-07-18 12:57:21
See also
AiiDA documentation: Quick installation
If aiida-crystal17 is installed, the crystal17.main computation
should be available:
In:
!verdi plugin list aiida.calculations crystal17.main
Inputs
basissets: required BasisSetData Use a node for the basis set of one of the elements in the structure. You h ...
code: required Code The Code to use for this job.
parameters: required CryInputParamsData the input parameters to create the .d12 file content.
structure: required StructureData structure used to construct the input fort.34 (gui) file
kinds: optional KindData additional structure kind specific data (e.g. initial spin)
metadata: optional
symmetry: optional SymmetryData the symmetry of the structure, used to construct the input .gui file (fort. ...
wf_folder: optional RemoteData An optional working directory, of a previously completed calculation, conta ...
Outputs
remote_folder: required RemoteData Input files necessary to run the process will be stored in this folder node ...
results: required Dict the data extracted from the main output file
retrieved: required FolderData Files that are retrieved by the daemon will be stored in this node. By defa ...
optimisation: optional TrajectoryData atomic configurations, for each optimisation step
structure: optional StructureData the structure output from the calculation
symmetry: optional SymmetryData the symmetry data from the calculation
Exit codes
1: The process has failed with an unspecified error.
2: The process failed with legacy failure mode.
10: The process returned an invalid output.
11: The process did not register a required output.
200: The retrieved folder data node could not be accessed.
210: The main (stdout) output file was not found
211: The temporary retrieved folder was not found
300: An error was flagged trying to parse the crystal exec stdout file
301: An error occurred parsing the 'opta'/'optc' geomerty files
350: The input file could not be read by crystal
351: Crystal could not find the required wavefunction file
400: The calculation stopped prematurely because it ran out of walltime.
401: The calculation stopped prematurely because it ran out of memory.
402: The calculation stopped prematurely because it ran out of virtual memory.
411: Scf convergence did not finalise (usually due to reaching step limit)
412: Geometry convergence did not finalise (usually due to reaching step limit)
413: An error encountered usually during geometry optimisation
414: An error was encountered during an scf computation
415: An unknown error was encountered, causing the mpi to abort
499: The main crystal output file flagged an unhandled error
510: Inconsistency in the input and output symmetry
520: Primitive symmops were not found in the output file
To use the python interface, first ensure a profile is loaded in the python kernel, and import the required modules:
In:
from aiida import load_profile
profile = load_profile()
profile.name
Out:
'test_crystal17'
In:
import os
from aiida.orm import Code
from aiida.plugins import (
DataFactory, WorkflowFactory, CalculationFactory)
from aiida.engine import run_get_node
from aiida_crystal17.common import display_json
from aiida_crystal17.tests import TEST_FILES
from aiida.tools.visualization import Graph
from jsonextended import edict
Input Node Creation¶
See also
Code¶
See also
AiiDA documentation: Setup a code
An Code node should be set up in
advance, to use the crystal17.basic calculation plugin, and call the
runcry17 executable (or mock_runcry17 used here for test
purposes).
In:
from aiida_crystal17.tests.utils import get_or_create_local_computer, get_or_create_code
computer = get_or_create_local_computer('work_directory', 'localhost')
code = get_or_create_code('crystal17.main', computer, 'mock_crystal17')
code.get_full_text_info()
Out:
[['PK', 23],
['UUID', '8451bd5e-397a-4014-9982-340b571a48c8'],
['Label', 'crystal17.main-mock_crystal17@localhost'],
['Description', ''],
['Default plugin', 'crystal17.main'],
['Type', 'remote'],
['Remote machine', 'localhost'],
['Remote absolute path', '//anaconda/envs/aiida-workshop/bin/mock_crystal17'],
['Prepend text', 'No prepend text'],
['Append text', 'No append text']]
Input Parameters (Geometry Independent)¶
The CryInputParamsData
supplies (geometry independent) data required to create the
input.d12 file.
In:
param_dict = {"scf":{"k_points": (8, 8)}}
params = DataFactory('crystal17.parameters')(data=param_dict)
params
Out:
<CryInputParamsData: uuid: 552ca25b-109f-4c25-8513-b441ba1b5d7c (unstored)>
The input data is validated against a JSON schema, which can be obtained
from the data_schema attribute.
Note
The only mandated key is scf.k_points (known as SHRINK in
CRYSTAL17)
In:
param_cls = DataFactory('crystal17.parameters')
edict.pprint(params.data_schema, keycolor="blue")
$schema: http://json-schema.org/draft-04/schema#
additionalProperties: False
description: Allowed Inputs For CRYSTAL17 .d12 file
properties:
basis_set:
additionalProperties: False
description: Basis sets input and control
properties: {...}
title: Block 2
type: object
geometry:
additionalProperties: False
description: Geometry input, manipulation and optimisation control
properties: {...}
title: Block 1
type: object
scf:
additionalProperties: False
dependencies: {...}
description: Single particle Hamiltonian and SCF control
properties: {...}
required: [k_points]
title: Block 3
type: object
title:
description: the title of the run
type: string
required: [scf]
title: CRYSTAL17 Input
type: object
In:
params = DataFactory('crystal17.parameters')(data={"scf": {}})
---------------------------------------------------------------------------
ValidationError Traceback (most recent call last)
<ipython-input-10-4a25c6adca90> in <module>()
----> 1 params = DataFactory('crystal17.parameters')(data={"scf": {}})
/Users/cjs14/GitHub/aiida-crystal17/aiida_crystal17/data/input_params.py in __init__(self, data, unflatten, **kwargs)
46 if unflatten:
47 data = unflatten_dict(data)
---> 48 self.set_data(data)
49
50 def _validate(self):
/Users/cjs14/GitHub/aiida-crystal17/aiida_crystal17/data/input_params.py in set_data(self, data)
64
65 # first validate the inputs
---> 66 self.validate_parameters(data)
67
68 # store all but the symmetry operations as attributes
/Users/cjs14/GitHub/aiida-crystal17/aiida_crystal17/data/input_params.py in validate_parameters(cls, dct)
29
30 """
---> 31 validate_against_schema(dct, cls.data_schema)
32
33 def __init__(self, data=None, unflatten=False, **kwargs):
/Users/cjs14/GitHub/aiida-crystal17/aiida_crystal17/validation/__init__.py in validate_against_schema(data, schema)
95 "\n".join(["- {} [key path: '{}']".format(
96 error.message, "/".join([str(p) for p in error.path]))
---> 97 for error in errors])
98 )
99
ValidationError: - 'k_points' is a required property [key path: 'scf']
It is also possible to reverse engineer the input data, from an existing
input file, using
extract_data.
In:
from aiida_crystal17.parsers.raw.inputd12_read import extract_data
param_dict, basis_sets, atom_props = extract_data("""\
MgO bulk
EXTERNAL
OPTGEOM
FULLOPTG
END
END
12 3
1 0 3 2. 0.
1 1 3 8. 0.
1 1 3 2. 0.
8 2
1 0 3 2. 0.
1 1 3 6. 0.
99 0
END
DFT
B3LYP
SPIN
END
SHRINK
8 8
ANDERSON
SMEAR
0.1
ATOMSPIN
2
1 1 2 -1
PPAN
END
""")
param_dict
Out:
{'title': 'MgO bulk',
'geometry': {'optimise': {'type': 'FULLOPTG'}},
'scf': {'dft': {'xc': 'B3LYP', 'SPIN': True},
'k_points': (8, 8),
'fock_mixing': 'ANDERSON',
'numerical': {'SMEAR': 0.1},
'post_scf': ['PPAN']}}
In:
from aiida_crystal17.parsers.raw.inputd12_write import write_input
print(write_input(param_dict, basis_sets, atom_props))
MgO bulk
EXTERNAL
OPTGEOM
FULLOPTG
ENDOPT
END
12 3
1 0 3 2. 0.
1 1 3 8. 0.
1 1 3 2. 0.
8 2
1 0 3 2. 0.
1 1 3 6. 0.
99 0
END
DFT
B3LYP
SPIN
END
SHRINK
8 8
ATOMSPIN
2
1 1
2 -1
SMEAR
0.1
ANDERSON
PPAN
END
Atomic Structure¶
The structure refers to a standard StructureData
node, and is used to create the main.gui.
Structures consist of:
A cell with a basis vectors and whether it is periodic, for each dimension
Sitewith a cartesian coordinate and reference to a kindKindwhich details the species and composition at one or more sites
The simplest way to create a structure is via ase:
In:
from ase.spacegroup import crystal
atoms = crystal(
symbols=[12, 8],
basis=[[0, 0, 0], [0.5, 0.5, 0.5]],
spacegroup=225,
cellpar=[4.21, 4.21, 4.21, 90, 90, 90])
struct_cls = DataFactory('structure')
structure = struct_cls(ase=atoms)
structure
Out:
<StructureData: uuid: 45ad9994-71b4-4ec3-8308-5f2bdb94f666 (unstored)>
These structures can be visualised using standard ASE methods.
In:
%matplotlib inline
import matplotlib.pyplot as plt
from ase.visualize.plot import plot_atoms
atoms = structure.get_ase()
fig, ax = plt.subplots()
plot_atoms(atoms.repeat((2,2,2)),
ax, radii=0.8, show_unit_cell=True,
rotation=('45x,0y,0z'));
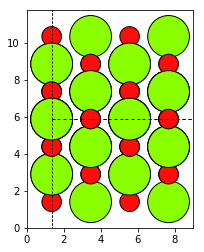
Structure visualisation, using ASE and Matplotlib.¶
As default, one kind is created per atomic species (named as the atomic symbol):
In:
structure.get_site_kindnames()
Out:
['Mg', 'Mg', 'Mg', 'Mg', 'O1', 'O1', 'O1', 'O1']
However, we may want to specify more than one kind per species (for example to setup anti-ferromagnetic spin). We can achieve this by tagging the atoms:
In:
atoms_afm = atoms.copy()
atoms_afm.set_tags([1, 1, 2, 2, 0, 0, 0, 0])
structure_afm = struct_cls(ase=atoms_afm)
structure_afm.get_site_kindnames()
Out:
['Mg1', 'Mg1', 'Mg2', 'Mg2', 'O', 'O', 'O', 'O']
..note:
::
Since we always use the
EXTERNALkeyword for geometry, any manipulation to the geometry is undertaken before calling CRYSTAL (i.e. we delegate the responsibility for geometry away from CRYSTAL).
Kind Specific Parameters¶
Also, we may want to add atom specific inputs to the .d12, such as
initial spin and frozen atoms (for optimisation).
In:
kind_cls = DataFactory("crystal17.kinds")
kind_data = kind_cls(data={
"kind_names": ["Mg1", "Mg2", "O"],
"spin_alpha": [True, False, False],
"spin_beta": [False, True, False],
"fixed": [False, False, True],
"ghosts": [False, False, False]
})
display_json(kind_data.kind_dict)
{
"Mg1": {
"spin_alpha": true,
"spin_beta": false,
"fixed": false,
"ghosts": false
},
"Mg2": {
"spin_alpha": false,
"spin_beta": true,
"fixed": false,
"ghosts": false
},
"O": {
"spin_alpha": false,
"spin_beta": false,
"fixed": true,
"ghosts": false
}
}
In:
from aiida_crystal17.parsers.raw.inputd12_write import create_atom_properties
atom_props2 = create_atom_properties(structure_afm, kind_data)
atom_props2
Out:
{'spin_alpha': [1, 2],
'spin_beta': [3, 4],
'unfixed': [1, 2, 3, 4],
'ghosts': []}
In:
print(write_input(param_dict, basis_sets, atom_props2))
MgO bulk
EXTERNAL
OPTGEOM
FULLOPTG
FRAGMENT
4
1 2 3 4
ENDOPT
END
12 3
1 0 3 2. 0.
1 1 3 8. 0.
1 1 3 2. 0.
8 2
1 0 3 2. 0.
1 1 3 6. 0.
99 0
END
DFT
B3LYP
SPIN
END
SHRINK
8 8
ATOMSPIN
4
1 1
2 1
3 -1
4 -1
SMEAR
0.1
ANDERSON
PPAN
END
Basis Sets¶
Basis sets are stored as separate
BasisSetData nodes, in a
similar fashion to UpfData. They
are created individually from a text file, which contains the content of
the basis set and (optionally) a YAML style header section, fenced by
---:
In:
mg_basis_path = os.path.join(TEST_FILES, 'basis_sets', 'sto3g', 'sto3g_Mg.basis')
with open(mg_basis_path) as handle:
print(handle.read())
---
author: John Smith
year: 1999
class: sto3g
---
12 3
1 0 3 2. 0.
1 1 3 8. 0.
1 1 3 2. 0.
The attributes of the basis set are stored in the database, and the md5 hash-sum is used to test equivalence of two basis sets.
In:
basis_cls = DataFactory('crystal17.basisset')
mg_basis, created = basis_cls.get_or_create(mg_basis_path)
display_json(mg_basis.attributes)
mg_basis.content
{
"md5": "0731ecc3339d2b8736e61add113d0c6f",
"year": 1999,
"class": "sto3g",
"author": "John Smith",
"element": "Mg",
"filename": "sto3g_Mg.basis",
"basis_type": "all-electron",
"num_shells": 3,
"atomic_number": 12
}
Out:
'12 3\n1 0 3 2. 0.\n1 1 3 8. 0.\n1 1 3 2. 0.'
A simpler way to create and refer to basis sets, is via a family group. All basis sets in a folder can be read and saved to a named family by:
In:
nfiles, nuploaded = basis_cls.upload_basisset_family(
os.path.join(TEST_FILES, 'basis_sets', 'sto3g'),
"sto3g", "group of sto3g basis sets",
extension=".basis", stop_if_existing=False)
basis_cls.get_basis_group_map("sto3g")
Out:
{'Mg': <BasisSetData: uuid: 832e1641-6c54-49c6-9039-42416fe8df66 (pk: 24)>,
'Ni': <BasisSetData: uuid: f9138f14-401b-4287-800e-8e6e4a95f0d6 (pk: 25)>,
'O': <BasisSetData: uuid: 3ab50757-05b6-48de-bd8e-6488472c3c69 (pk: 26)>}
or at the command line:
In:
!verdi data crystal17.basis uploadfamily --help
Usage: verdi data crystal17.basis uploadfamily [OPTIONS]
Upload a family of CRYSTAL Basis Set files.
Options:
--path PATH Path to a folder containing the Basis Set
files
--ext TEXT the file extension to filter by
--name TEXT Name of the BasisSet family [required]
-D, --description DESCRIPTION A description for the family
--stop-if-existing Abort when encountering a previously uploaded
Basis Set file
--dry-run do not commit to database or modify
configuration files
-h, --help Show this message and exit.
In:
!verdi data crystal17.basis listfamilies
Family Num Basis Sets
-------- ----------------
sto3g 3
Basis families can be searched by the elements they contain:
In:
basis_cls.get_basis_groups(["Ni", "O"])
Out:
[<Group: "sto3g" [type crystal17.basisset], of user chrisj_sewell@hotmail.com>]
Basis sets can also be extracted for a particular structure.
In:
basis_cls.get_basissets_from_structure(structure, "sto3g")
Out:
{'Mg': <BasisSetData: uuid: 832e1641-6c54-49c6-9039-42416fe8df66 (pk: 24)>,
'O': <BasisSetData: uuid: 3ab50757-05b6-48de-bd8e-6488472c3c69 (pk: 26)>}
Important
Unlike aiida-quantumespresso.pw,
crystal17.main uses one
basis sets per atomic number only NOT per kind.
This is because,
using multiple basis sets per atomic number is rarely used in
CRYSTAL17,
and is limited anyway to only two types per atomic number.
Symmetry¶
In the main.gui file, as well as using the dimensionality
(i.e. periodic boundary conditions), basis vectors and atomic positions,
provided by the structure, we also need to specify the atomic
symmetry of the structure.
SymmetryData is used to store
this data, as a validated dictionary.
Note
The operations are given as a flattened version of the rotation
matrix,
followed by the translation vector.
In:
symmetry_cls = DataFactory("crystal17.symmetry")
edict.pprint(symmetry_cls.data_schema, keycolor="blue")
$schema: http://json-schema.org/draft-07/schema
additionalProperties: True
properties:
basis:
description: whether the symmetry operations are fractional or cartesian
enum: [fractional, cartesian]
type: string
computation:
description: details of the computation
type: object
equivalent_sites:
description: mapping table to equivalent atomic sites
items: {...}
type: array
hall_number:
description: Hall number defining the symmetry group
maximum: 530
minimum: 1
type: [null, integer]
operations:
description: symmetry operations, should at least include the unity
operation
items: {...}
minItems: 1
type: array
uniqueItems: True
required: [hall_number, operations, basis]
title: structure symmetry settings
type: object
The simplest symmetry would be the unitary operator.
In:
symmetry = symmetry_cls(data={
"hall_number": 1,
"basis": "fractional",
"operations": [
[1,0,0,0,1,0,0,0,1,0,0,0]
]
})
symmetry.attributes
Out:
{'hall_number': 1, 'basis': 'fractional', 'num_symops': 1}
The full symmetry operations of a periodic structure, can be computed
using the crystal17.sym3d workflow. This uses the
spglib <https://atztogo.github.io/spglib/>_ library to compute
symmetries, but with the added constraint that sites with the same
Kind must be symmetrically equivalent.
In:
!verdi plugin list aiida.workflows crystal17.sym3d
Inputs
settings: required Dict
cif: optional CifData
metadata: optional
structure: optional StructureData
Outputs
symmetry: required SymmetryData
structure: optional StructureData
Exit codes
1: The process has failed with an unspecified error.
2: The process failed with legacy failure mode.
10: The process returned an invalid output.
11: The process did not register a required output.
300: One of either a structure or cif input must be supplied
301: The supplied structure must be 3d (i.e. have all dimensions pbc=true)"
302: Idealize can only be used when standardize=true
303: The kind names supplied are not compatible with the structure
304: Error creating new structure
305: Error computing symmetry operations
In:
sym3d_cls = WorkflowFactory("crystal17.sym3d")
builder = sym3d_cls.get_builder()
builder.settings = {"symprec": 0.01}
builder.structure = structure
sym_result = run_get_node(builder)
In:
graph = Graph(graph_attr={'size': "8,8!", "rankdir": "LR"})
graph.recurse_ancestors(sym_result.result["symmetry"],
annotate_links="both")
graph.graphviz
Out:

crystal17.sym3dworkflow provenance graph.¶
This workflow can also optionally compute the primitive and/or standardised form of the structure, before computing the symmetry of the new structure.
In:
builder.settings = {
"symprec": 0.01,
"compute_primitive": True,
"standardize_cell": True}
sym_result2 = run_get_node(builder)
graph = Graph(graph_attr={'size': "9,9!", "rankdir": "LR"})
graph.recurse_ancestors(sym_result2.result["structure"],
annotate_links="both")
graph.recurse_ancestors(sym_result2.result["symmetry"],
annotate_links="both")
graph.graphviz
Out:

crystal17.sym3dworkflow provenance graph, including primitive cell
calculation.¶
The other option is to idealize the structure, which removes
distortions of the unit cell’s atomic positions, compared to the ideal
symmetry.
Setting Up and Running the Calculation¶
See also
AiiDA documentation: Processes
CryMainCalculation
provides a helper function to create, populate and validate the input
builder.
In:
from aiida_crystal17.tests import get_test_structure_and_symm
from aiida_crystal17.data.kinds import KindData
calc_cls = CalculationFactory('crystal17.main')
structure, symmetry = get_test_structure_and_symm("NiO_afm")
kind_data = KindData(data={
"kind_names": ["Ni1", "Ni2", "O"],
"spin_alpha": [True, False, False],
"spin_beta": [False, True, False]})
calc_builder = calc_cls.create_builder(
parameters={
"title": "NiO Bulk with AFM spin",
"scf.single": "UHF",
"scf.k_points": (8, 8),
"scf.spinlock.SPINLOCK": (0, 15),
"scf.numerical.FMIXING": 30,
"scf.post_scf": ["PPAN"]
},
unflatten=True,
structure=structure,
symmetry=symmetry,
kinds=kind_data,
bases="sto3g",
code=Code.objects.get(label="crystal17.basic-mock_crystal17@localhost"),
metadata={"options": {"resources": {
"num_machines": 1, "num_mpiprocs_per_machine": 1}}}
)
display_json(calc_builder)
{
"metadata": {
"options": {
"resources": {
"num_machines": 1,
"num_mpiprocs_per_machine": 1
}
}
},
"basissets": {
"Ni": [
"uuid: f9138f14-401b-4287-800e-8e6e4a95f0d6 (pk: 25)"
],
"O": [
"uuid: 3ab50757-05b6-48de-bd8e-6488472c3c69 (pk: 26)"
]
},
"parameters": [
"uuid: 92bba44f-b3df-4e04-ad69-a6b411cf787b (unstored)"
],
"structure": [
"uuid: 8d626706-24f9-43b8-bbf3-9b01b851e591 (pk: 169)"
],
"symmetry": [
"uuid: 2b594d60-b846-4a6b-aeb1-b0894984d438 (pk: 171)"
],
"kinds": [
"uuid: ed253aa1-bac0-4da6-8d00-679cf23e7dae (unstored)"
],
"code": [
"Remote code 'crystal17.basic-mock_crystal17@localhost' on localhost,",
"pk: 1, uuid: d9ae48df-22cf-4ee2-9d47-0e16f48c8b0a"
]
}
In order to run the computation, the builder can be parsed to one of the
AiiDA run (blocking execution) or submit (non-blocking
execution) functions:
In:
result, calcnode = run_get_node(calc_builder)
The process can be monitored on the command line:
In:
!verdi process list -a -D desc -l 4
PK Created Process label Process State Process status
---- --------- --------------------- --------------- ----------------
174 19s ago CryMainCalculation ⏹ Finished [0]
170 22s ago compute_symmetry ⏹ Finished [0]
168 23s ago primitive_structure ⏹ Finished [0]
167 23s ago Symmetrise3DStructure ⏹ Finished [0]
Total results: 4
Info: last time an entry changed state: 5s ago (at 12:33:22 on 2019-07-18)
Once the calculation is complete, a CalcJobNode will be created, to
store the settings and outcome of the computation. Crucially, if the
computation has completed successfully, the exit_status will be
0.
This can be assessed on the command line or with the python API.
In:
!verdi process show {calcnode.pk}
Property Value
------------- ------------------------------------
type CalcJobNode
pk 174
uuid 99770bbe-faed-45ac-bf27-ded8191ea560
label
description
ctime 2019-07-18 12:33:08.329202+00:00
mtime 2019-07-18 12:33:22.196416+00:00
process state Finished
exit status 0
computer [1] localhost
Inputs PK Type
---------- ---- ------------------
basissets
Ni 25 BasisSetData
O 26 BasisSetData
code 1 Code
kinds 173 KindData
parameters 172 CryInputParamsData
structure 169 StructureData
symmetry 171 SymmetryData
Outputs PK Type
------------- ---- ----------
remote_folder 175 RemoteData
results 177 Dict
retrieved 176 FolderData
In:
print(calcnode.is_finished_ok)
print(calcnode.process_state)
print(calcnode.exit_status)
True
ProcessState.FINISHED
0
If the calculation fails, there are three things that should be checked:
The calculation’s exit_message
The calculation’s log messages and scheduler output
The
resultsoutput node (if available)
In:
print("Exit Message:", calcnode.exit_message)
from aiida.cmdline.utils.common import get_calcjob_report
print(get_calcjob_report(calcnode))
Exit Message: None
*** 174: None
*** (empty scheduler output file)
*** (empty scheduler errors file)
*** 0 LOG MESSAGES
In:
!verdi process report {calcnode.pk}
*** 174: None
*** (empty scheduler output file)
*** (empty scheduler errors file)
*** 0 LOG MESSAGES
Analysis of Outputs¶
The Graph can be used to
visualise the calculations provenance graph:
In:
graph = Graph(graph_attr={'size': "6,8!", "rankdir": "LR"})
graph.add_node(calcnode)
graph.add_incoming(calcnode, annotate_links="both")
graph.add_outgoing(calcnode, annotate_links="both")
graph.graphviz
Out:

crystal17.main calculation provenance graph (SCF only).¶
The retrieved FolderData output node contains the CRYSTAL17 main
input and output file.
In:
calcnode.outputs.retrieved.list_object_names()
Out:
['_scheduler-stderr.txt', '_scheduler-stdout.txt', 'fort.34', 'main.out']
The results Dict output node contains key values extracted from
the CRYSTAL17 main output file.
In:
display_json(calcnode.outputs.results.attributes)
{
"units": {
"angle": "degrees",
"energy": "eV",
"length": "angstrom",
"conversion": "CODATA2014"
},
"energy": -85124.893667339,
"errors": [],
"header": {
"crystal_version": 17,
"crystal_subversion": "1.0.1"
},
"volume": 36.099581472,
"warnings": [],
"calculation": {
"n_ao": 46,
"spin": true,
"type": "unrestricted open shell",
"n_atoms": 4,
"n_shells": 14,
"n_symops": 16,
"n_core_el": 40,
"n_electrons": 72,
"n_kpoints_ibz": 75,
"n_kpoints_gilat": 75
},
"energy_units": "eV",
"parser_class": "CryMainParser",
"parser_errors": [],
"mulliken_spins": [
3.057,
-3.057,
-0.072,
0.072
],
"parser_version": "0.9.0b5",
"scf_iterations": 13,
"number_of_atoms": 4,
"parser_warnings": [],
"mulliken_charges": [
0.398,
0.397,
-0.398,
-0.397
],
"parser_exceptions": [],
"mulliken_electrons": [
27.602,
27.603,
8.398,
8.397
],
"mulliken_spin_total": 0.0,
"number_of_assymetric": 4,
"execution_time_seconds": 187
}